Глюконеогенез – процесс превращения амино-кислот и глицерина в глюкозу. С ростом популярности кето-диеты хочется уделить время тому, почему кето – не волшебная диета, а имеет ряд ограничений.
Если сумма потенциальных энергий конечных продуктов реакции сильно меньше чем изначальных реактантов, то реакцию принято считать необратимой. У нашего организма есть обходные пути синтеза нужных молекул, но это энергонеэффективный процесс. Для восстановления молекулы глюкозы организм затратит заметно больше энергии, чем может извлечь из нее. Зачем же нам тогда глюкоза?
Я уже касался этой темы более подробно в заметке про глюкозу и мозг. Жировые кислоты могут быть использованы только для получения энергии и ее запасания в виде жира. Глюкоза – это еще и строительные углеродные блоки для поддержания целостности белкового тела, и источник (через пентозо-фосфатный путь) NADPH для восстановительных реакций и рибозы (строительного блока нуклеиновых кислота).
Наивно думать, на мой взгляд, что склонять организм к энергонеэффективному процессу продолжительное время можно без последствий.
Глюконеогенез и катехоламины
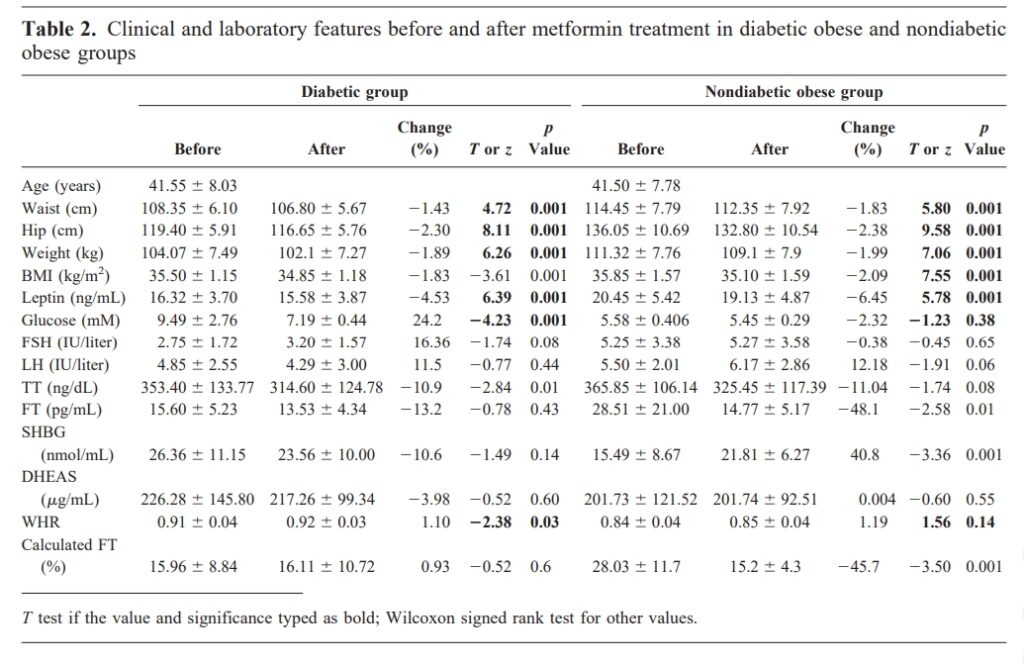
Оксалоацетат – промежуточное звено цикла Кребса. Проблема в том, что оно истощается не только самим циклом лимонной кислоты, но и глюконеогенезом, и синтезом амино-кислот.
Еще в 1946-м году Ленингер (Lehninger) показал, что рост кетоновых тел обратно-пропорционален уровню оксало-ацетата [1].
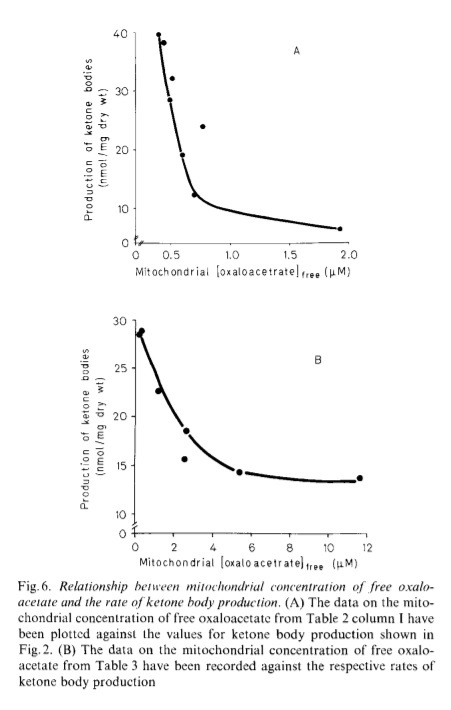
Больше кетонов значит ниже глюкоза, значит активнее должен протекать глюконеогенез.
Преобразование пирувата в оксалоацетат (ступенька восстановления глюкозы, сам пируват несложно получить из амино-кислот и других компонентов цикла Кребса) катализирует пируват карбоксилаза. Пируват карбоксилазу а кортикостероиды, глюкагон (считайте дефицит глюкозы) и катехоламины.
Катехоламины – это дофамин, адреналин, норадреналин. Сигналы, посылаемые нервной системой. В принципе это способ нашей нервной системы «поддать газу» на глюконеогенез.
Кето-диета имитирует голод. На молекулярном уровне в том числе. Она активирует симпато-адренальную систему [3, 4]. Проще говоря, активируется часть нервной системы, отвечающая за стресс.
Предполагаю, что своеобразное и очень интересное ментальное состояние на кето – результат работы не только кетоновых тел, но и катехоламинов. Также напрашивается частичная аналогия с некоторыми антипичными антидепрессантами вроде бупропиона.
Глюконеогенез и цАМФ
Хронический глюконеогенез будет связан с цАМФ. Циклические молекулы в биохимии являются частью системы second messengers. При недостатке глюкозы поджелудочная будет выбрасывать вместо инсулина глюкагон. Гормон – экстраклеточный сигнал. А second messengers – это клеточные молекулы, активируемые внешним сигналом, которые амплифицируют (усиливают) изначальный сигнал и проделывают еще какую-то работу.
цАМФ связана с модулированием функции Т клеток [5].
Циклические молекулы (цГМФ и цАМФ) приводят к высвобождению гладкой мускулутарой кальция. И в последующем к эрекции. Повышенный уровень цАМФ на кето могут помочь людям, чья невозможно поддержать эрекцию связана с функцией цГМФ. Анекдотические случаи уже есть на кето.
Однако циклические молекулы имеют и обратную сторону. Они могут при некоторых обстоятельствах приводить к сужению сосудов [6] (а не только способствовать их расширению и улучшению микроциркуляции).
В целом роль цАМФ скорее положительная. Например, сдвиг митохондрий в сторону окислительного метаболизма [7]. Но роль цАМФ многогранна, а в рамках дилеммы плохо/хорощо двояка.
Метформино-подобный эффект кето и тестостерон
В свое время я показывал, что кето-диета приводит к обратимой деградации комплекса I митохондрий. Что схоже с эффектом метформина и даже имеет с ним синергетичный эффект.
Это может быть полезно для ряда метаболических заболеваний. Но довольно вредно для фертильности и уровня анаболических гормонов. Снижение тестостерона на метформине – известная проблема.
Отсюда большое противоречение про кето и тестостерон. «Больше холестерина – больше тестостерона» – несколько нерабочий слоган. Клетки Лейдига с подавленным 1-ым комплексом будут производить больше тестостерона или меньше? Даже при избытке холестерина.
Про митоходрии, близость и работу комплексов и важности всего этого для фертильности я уже писал.
Нормальные клетки с эффектом Варбурга и кето
Эффект Варбурга – это предпочтение гликолиза даже в условиях избытка кислорода – один из метаболических признаков рака.
Проблема в том, что ряд нормальных клеток полностью или в основном гликолитичны: клетки Мюллера (глаза), стволовые клетки, лейкоциты, часть клеток выделительной системы.
При избытках кислородных радикалов стволовые клетки дифференциируются (+/- контекстуальны). И отчасти по этой причине есть известное противоречие кето и рака почек / мочевыводящих путей.
Значит эффект Варбурга то, что к кето надо подходить осмысленно. Четко понимая каких целей мы хотим достигнуть и какие риски нас могут ждать.
Кето и выведение метаболических кислот
Кетоны – метаболические кислоты. Кислотность (водород) от углеводов выводится легкими за минуты. Кетонов и жиров – почками в течение недель. Подробнее в одной из прошлых заметок.
Опуская то, что вы можете прочитать в другой заметке, кето снижает pH почти. В 30х (до эры антибиотиков) кето использовали для снижения pH мочи, для эрадикации бактерий урогенитального тракта [9].
В России нет теста крови на бикарбонат. Мы не может контролировать Anion Gap и метаболический ацидоз через кровь (организм от него будет восстанавливаться неделями). Нужно контролировать pH мочи. При необходимости добавляя источники бикарбонатов.
Кето и ЖКТ
Желчь выбрасывается при поедании жирной пищи. С удаленной желчью кето – норма. Такая глубина анализа подходит, если у вас сектантское мышление или если вам недавно провели интернет, но еще не научили искать информацию и критически ее анализировать.
Кето – метаболическая кислота. Гастроэнтерологи советуют завтрак овсянкой как способ уменьшить химический урон стенках желудках, на которые и придется основной урон от дисфункций связки печень – желчный пузырь.
Кето ухудшает симптоматику ГЭРБ.
Кето приводит к росту билирубина. Привет людям с болезнью Жильбера
И многое другое. Суммировал информацию традиционно Джон Бриссон.
When Excess Fat in the Diet Can Cause Digestive Issues and How to Improve Fat Digestion
Ketogenic Diets, Effects On Your Gut and Health and How To Follow Them
Кето, выводы и примеры виртуальных пациентов
Кето-диета имитирует голодание. На очень многих уровнях. И всегда ли долгосрочное псевдо-голодание хорошо? Конечно же нет.
- Кето (глюконеогенез) приводит к росту катехоламинов; Может привести к симпатическому тонусу.
- Роль цАМФ (глюконеогенез) неоднозначна;
- Кето приводит к обратимому подавлению комплекса I и метформино-подобному эффектов, в том числе снижению тестостерон;
- У нас есть ряд почти гликолитичных клеток, чью функцию кето не улучшит, но за счет сдвига в сторону окисления жиров и за счет снижения pH может иметь негативное влияние;
- Кетоны – метаболические кислоты, снижают pH мочевыделительной системы, увеличивают на нее нагрузку на недели вперед;
- Кетоны – метаболические кислоты, что является дополнительным фактором риска при целом ряде проблем с ЖКТ.
Виртуальные примеры
Пример 1. У нас пациент с депрессией после развода, который не реагирует на СИОЗС, но хорошо реагирует на бупропион (поднимает дофамин и норадреналин). Кето-диета может полноценно заменить антидепрессант. При дополнении кето поведенческой терапией и другой поддерживающей терапией (d3, окситоцин интраназально итд) может спасти пациента от нежелательных явлений/
Пример 2. У нас пациент с явными митохондриальными дефектами. У нас в арсенале кето и гипер/гипо окситерапии, который помогут уничтожить дефектные митоходрии. Но нам обязательно нужно дополнить подобную терапию интервенциями, которые приведут к росту митохондрий (PQQ. Тренировки тела/ума на это итд). Или мы можем рассмотреть молекулы переносящие электрон с 1 на 3 и с 1 на 4 комплексы (ибеденон, метиленовый синий).
Пример 3. Пациент с 4-й месяц кето говорит о симптомах симпатического тонуса. Плюс пациент начинает быстрее свирепеть. При курильщиках, например. Возможно, проблема в сужении сосудов из-за катехоламинов.
Пример 4. У пациента семейная история болезней желчного пузыря и синдром Жильбера. На кето вырастит билирубин и появятся/усилятся симптомы синдрома. Кето может усилить/актуализировать проблему «перетяжки» желчного пузыря, приведя к ГЭРБ и повреждению желудка. Вероятно, стоит продумать поддерживающие терапии или отказаться от кето (в зависимости от целей/пациента).
Пример 6 Мужчина с хроническим простатитом. Кето снизит pH в мочевыделительной системе, делая простату менее доступной для иммунных клеток (которые тоже белковые тела и их функция зависит от pH), кето может негативно повлиять на симптоматику за счет сужения сосудов и, возможно, неоднозначной роли на гликолитичные клетки.
Пациент 7. Девушка до 30, которая хочет забеременеть в первую очередь, во вторую – немного улучшить метаболическое здоровье. Скоро поедет отдыхать. Кето ей не нужно. Есть альтернативные стратегии.
Источники:
- Role of Free Oxaloacetate in Ketogenesis;
- Structure, function and regulation of pyruvate carboxylase;
- Effect of low-carbohydrate-ketogenic diet on metabolic and hormonal responses to graded exercise in men
- 34-day total fast in an adult man
- Cyclic adenosine monophosphate is a key component of regulatory T cell–mediated suppression
- cGMP and cAMP cause pulmonary vasoconstriction in the presence of hemolysate
- Cyclic AMP signaling in cardiac myocytes
- Long-term High Fat Ketogenic Diet Promotes Renal Tumor Growth in a Rat Model of Tuberous Sclerosis
- The Ketogenic Diet in Bacilluria